Jul 9, 2019 |
So we’ve been writing a lot about the importance of using genomic data in Clinical Decision Support (CDS). We’ve also written about the importance of Genomic Archiving and Communication Systems (GACS) to store the petabytes upon petabytes of data that will inevitably arise from genomic sequencing. What we haven’t touched on yet is a rather uncomfortable truth – a good portion of the identified genomic variants that come back from genetic tests can actually be false positives according to some reports . How do we know which variants are safe to use in CDS and which ones to ignore?
Right now, if a particular variant is of interest, it can be put through a confirmatory testing process (e.g. via Sanger sequencing) to see if it’s real or a false positive. That is wholly inconvenient and not at all practical from a workflow perspective.
Imagine a patient, John Doe, who had whole genome sequencing performed five years ago as part of a cardiomyopathy evaluation. Of the 4 million variants found, those in a select panel of cardiac genes known to be pathogenic were reported back to the ordering cardiologist, after going through a rigorous CLIA-approved quality assurance process that includes confirmatory testing via Sanger sequencing to rule out false positives.
Two years later, the American College of Medical Genetics and Genomics issued recommendations for reporting of secondary findings in genome sequencing, and John Doe’s primary care provider wants to know if John has any of these potentially actionable variants. Around the same time, John Doe’s cardiologist recommends initiation of clopidogrel, but first wants to know if John has any potential drug-gene interactions.
When we look back at John’s DNA results from five years ago, we find a variant in the LDLR gene (associated with Familial Hypercholesterolemia) that was only recently discovered to be pathogenic, and we find a variant in CYP2C19 associated with significant reduction in clopidogrel activity. Both of these variants have been identified in John’s DNA using an industry-standard bioinformatics pipeline, but neither of these variants (at least so far) has had confirmatory testing via Sanger sequencing. So, should the LDLR and CYP2C19 variants be reported back to the primary care provider and cardiologist? Do we need to first ask the lab to go through the time and expense of confirmatory testing (which may require a new specimen)? And what if this were a real-time clinical decision support scenario – should we intervene before the cardiologist signs the clopidogrel order?
Here at Elimu, we are interested in genomics-based clinical decision support. Our model is one in which raw next-generation sequencing files reside in a GACS, and are served up on-demand to various applications (such as the EHR or a clinical decision support system) via a FHIR interface, as illustrated here:
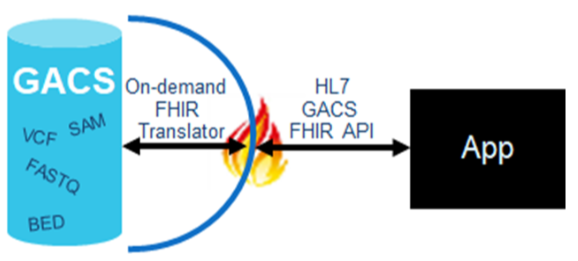
A challenge with this model however, is that the quality of variant calling is not uniform across the genome – there are false positives, there are false negatives, etc. So how can we know if an identified variant is safe for clinical use? Obviously this puts a little damper on real-time decision support if we have to invoke manual laboratory processes before we can alert the clinician to potentially relevant findings in a patient’s genome.
These decisions all hinge on our ability to objectively determine the analytic validity of variant calls. For the purposes of this post, we’ll define ‘analytic validity’ as they do in a recent National Academies of Sciences, Engineering, and Medicine report, which equates analytic validity with technical test performance (e.g. if a lab reports a variant, is it a true positive; if a lab does not identify a variant, is it a true negative). Analytic validity can be contrasted against ‘clinical validity’ (e.g. how well does the presence of a variant predict a disease) and ‘clinical utility’ (e.g. if you know there is a variant, and if you know the variant is highly predictive, can you improve outcomes).
Variant calling is a process whereby data obtained from a person’s next-generation sequencing is compared against reference DNA to look for differences, or variants. Per Dr. Pevsner, the steps involved are “extraordinarily complex”, with different software tools often coming up with different results. This complexity and discordance across platforms, along with a lack of objective metrics for assessing analytic validity, fuels the belief that confirmatory testing is typically warranted.
We were recently inspired by two articles, prompting us to write this article. The first, by J. Zheng and colleagues, concluded that “a new variant with high quality from a well‐validated capture‐based NGS workflow can be reported directly without validation”. The second, by S. Lincoln and colleagues, specifically examines when and if confirmatory testing is required. The article describes a validated algorithm that might be used to tier variants into different confidence groups. Figure 1 from the article provides an intuitive illustration of the output of the algorithm.
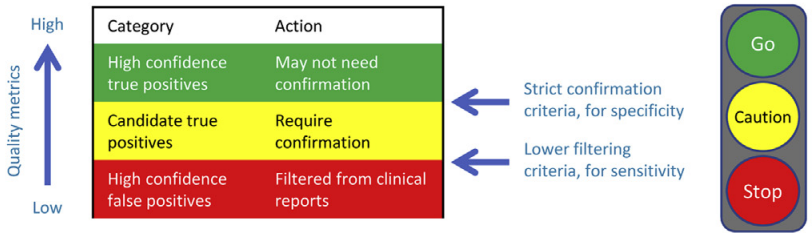
Using these confidence groups, a decision support engine might take this approach: If the variant in question is RED, it is likely a false positive, so ignore it; if the variant in question is GREEN, it is likely a true positive, and can be treated as such; if the variant in question is YELLOW, it may be a true positive, but tailor the recommendation (e.g. to indicate the need for confirmation).
Imagine, a given person’s DNA will differ from a reference DNA by around 4 million variants. If we can find a way to discern those variants that are safe to use clinically without the cost and effort of confirmatory testing, we open the flood-gates of genomic medicine, we enable life-long reanalysis of a person’s DNA, and we enable clinical care to be that much more precise.
“Ideally, a person’s genomic information should be available to themselves and their
health practitioners over their life course, enabling the underlying data to be reanalysed
as analytic tools and evidence for its use in care improves”. Gaff, 2017
That quote inspires us everyday to incorporate genomics data into CDS; specifically via GACS. As we continue down this path with great partners like AI2, who we are partnering with to build an open source GACS with FHIR interface for fellow innovators, we’ll begin to experiment with solutions like S. Lincoln’s to provide confidence levels to variant data so that everyone’s genomic information can be useful throughout their life course.